
AI-Driven Remote Patient Monitoring App for Chronic Care Management
- 20%
- Reduction in clinical decision-making time
- 150+
- Patients adopted in 12 weeks
- 100%
- HIPAA compliance
Clinical data sitting in spreadsheets across 12 sites. Regulatory submissions delayed because your systems can't produce the right audit trail. Field force training that takes 3 months to roll out.
We build clinical trial management systems, regulatory document platforms, HCP training tools, and pharmacovigilance systems with 21 CFR Part 11 compliance and GDPR designed in from the first sprint, not added when the regulatory review finds gaps.
Clinical trial management, drug tracking, and regulatory compliance systems built with 21 CFR Part 11 and GDPR in mind
HCP training platforms that cut field force onboarding from months to weeks
We built a pharma training platform that reduced onboarding time from 6 weeks to 10 days
100+ products shipped including healthcare and life sciences technology
What you can count on
Retention
3+ years
Average client relationship across active accounts
First milestone
Week 3
Working prototype in front of real users
Pricing
Fixed price
Scope and cost agreed before work starts
Team
No handoffs
The senior engineers who scope the build also ship it
The problem
Your trial data is spread across Excel files on 12 different laptops?
Regulatory submissions take twice as long as they should because your systems weren't built for compliance?
Short answer
RaftLabs builds custom pharmaceutical software for pharma companies replacing manual, error-prone processes. Clinical trial management systems, drug serialization platforms, regulatory compliance tools (21 CFR Part 11 and GDPR), patient management portals, and HCP training platforms. Most builds are production-ready in 12-16 weeks with audit trails and compliance documentation built in from sprint one.
Trusted by

Clinical trial data collection still using paper case report forms requiring manual transcription
Field investigators complete paper CRFs at clinical sites, then data entry staff manually key them into a central database. Transcription errors introduce data quality issues that require source data verification to resolve, adding weeks to data cleaning before a regulatory submission can be prepared. That's time and money lost before you've even reached the filing stage. We build electronic data capture systems where investigators enter data directly into structured eCRF screens with validation rules, cutting the transcription step and the errors it creates.
Regulatory submission dossier assembly taking months of manual document collation
Regulatory affairs teams preparing an IND, NDA, or MAA dossier spend months locating documents across shared drives, version-controlling them manually, and assembling them into CTD format. Submission timelines slip because document readiness is tracked in spreadsheets with no automated status visibility. Every day of delay is a day further from approval. According to the European Medicines Agency's internal data (as reported by Citeline's Pink Sheet, 2023), only 30-40% of Marketing Authorisation Applications submitted to the EMA between 2018 and 2022 arrived on time, with 42% of applicants requesting extensions due to insufficient data maturity. We build document management systems structured around the CTD module hierarchy, with version control, approval workflows, and submission package assembly built into the platform.
Pharmacovigilance adverse event tracking managed in fragmented systems with no automated signal detection
Adverse event reports arrive through multiple channels: call centres, clinical sites, literature review, and spontaneous reports. They get entered into separate systems with no automated aggregation. Signal detection relies on periodic manual review of case counts rather than automated pattern analysis across the full case database. Missed signals mean missed obligations. We build pharmacovigilance platforms that consolidate adverse event intake, route cases through medical review workflows, and surface signals from aggregate data without manual case counting.
Quality management system running on disconnected paper-based processes that auditors flag each inspection
SOPs, training records, deviation reports, CAPA records, and change controls exist in separate filing systems: some paper, some shared drive, with no single view of quality system status. FDA or EMA inspectors arrive and the QA team spends days locating records and demonstrating compliance. That cost in preparation time is avoidable. We build electronic quality management systems where SOPs, training, deviations, CAPAs, and change controls are managed in one platform with audit-ready reports generated on demand.
Clinical trial data drives regulatory submissions. It affects drug approval timelines. It has to be accurate, auditable, and accessible across multiple sites and time zones.
Most mid-size pharma companies run their trials across a mix of Excel, email, and paper case report forms. That works until it doesn't, and it stops working at the moment you need to file. We build eClinical systems that capture trial data at the source, enforce protocol compliance, and produce audit-ready outputs.
Best for
Drug serialization is no longer optional in most markets. DSCSA (US), FMD (EU), and similar regulations require unique identifiers on each saleable unit and the ability to trace any product from manufacturer to patient.
We build serialization systems and track-and-trace platforms that generate, apply, and report unique identifiers across your supply chain. These connect to your existing ERP or manufacturing execution system and produce the EPCIS reports your trading partners and regulators require.
Best for
Medical representatives need to learn complex clinical data, product positioning, and compliance rules before they talk to a single doctor. Traditional training, classroom sessions, printed manuals, in-person sign-offs, takes 6 to 10 weeks and doesn't scale.
We built a pharmaceutical training platform for a global pharma company that cut field force onboarding from 6 weeks to 10 days. The platform delivers digital modules, competency assessments, and certification tracking, with a full audit trail for regulatory sign-off.
Best for
Regulatory submission packages for FDA, EMA, PMDA, and other agencies are enormous, structured, and must be correct. Documents get submitted in CTD format, with cross-references, metadata, and electronic submission requirements that differ by region.
Manual document management in shared drives creates version control problems and delays submissions. We build document management systems for regulatory affairs teams that enforce structure, track versions, and produce submission-ready packages.
Best for
Clinical trials and disease management programs that require patient monitoring face a real data challenge: patients aren't at the clinic. Their vital signs, symptom reports, and medication adherence data must be captured remotely and fed into clinical records.
We've built remote patient monitoring platforms with medical-grade data handling, HIPAA-compliant storage, and integration with clinical data systems. Patients use a mobile app. Clinicians see structured data in their dashboard.
Best for
Adverse event reporting is a post-market obligation. Missing a report, submitting late, or producing an inaccurate CIOMS or MedWatch form can result in regulatory action.
We build pharmacovigilance tools that capture adverse events from multiple sources: call centres, clinical sites, literature, and spontaneous reports. Each case routes through a structured medical review and regulatory submission workflow.
Best for
Yes. For systems that require FDA compliance under 21 CFR Part 11, we design with the specific technical controls the regulation requires: audit trails with timestamps and user identification, electronic signature implementation with identity binding, system access controls, and data integrity validation. We can also support the validation documentation (IQ/OQ/PQ) that GxP-regulated clients need. We're not a validation consultancy, you'll need a qualified validation team to sign off, but we build systems that are designed to pass.
Yes. Our most direct reference is a pharmaceutical training platform we built for a global pharma company with a large field force. The platform delivered training content, tracked completions, and produced compliance certificates. We cut their field force onboarding time from 6 weeks to 10 days. We've also built patient monitoring systems with clinical-grade data handling and HIPAA-compliant architecture.
We can integrate with SAP, Oracle, Veeva, and most systems that offer an API. For legacy systems without a modern API, we've built integration layers using batch file exchange, database connectors, and middleware. The integration approach depends on what your existing system supports, and we scope that work as part of the discovery process.
For systems handling patient data, we design with data minimisation, encryption at rest and in transit, role-based access controls, and full audit trails. We use compliant cloud hosting (AWS or Azure in appropriate regions) and make sure data residency requirements are met. We're not a legal or compliance firm and don't give compliance advice, but we've built systems that passed their clients' HIPAA and GDPR reviews. We document our architecture decisions so your compliance team can review them.
A focused build, like an HCP training platform or an adverse event intake system, typically takes 12 to 16 weeks from kickoff to go-live. Clinical trial management systems are more complex, typically 4 to 6 months for a full build. Systems that require validation documentation take longer because the documentation process runs alongside development. We agree on milestones at the start so you know what to expect at each stage.
Clinical trial management software development
Drug serialization and track-and-trace software
HCP training platform development for pharma
Regulatory document management software for pharma
Remote patient monitoring software for pharma
Pharmacovigilance software development
What clients say
Three-year average engagement. Founders and operators describing the work in their own words. No marketing varnish.
PDC has been a great addition to our clinic. It helps us stay connected with patients and makes clinical decision-making significantly faster.
01 / 02
AI Document Intelligence
Extract structured data from regulatory submissions, clinical study reports, adverse event forms, and pharmacovigilance documents.
Business Process Automation
Automate submission workflows, label change management, PV case processing, and regulatory reporting.
AI Agent Development
Autonomous agents for literature surveillance, safety signal detection, and regulatory query response.
Custom Software Development
Custom eTMF, regulatory information management, and pharmacovigilance platforms built to your 21 CFR and EMA requirements.
Tell us your regulatory obligations, your clinical data workflows, and where your current systems create compliance risk or operational overhead. We'll scope the right platform and give you a fixed cost.
Stay on topic

Service
AI for Healthcare Organisations
See the service
Service
Teletherapy Platform Development
See the service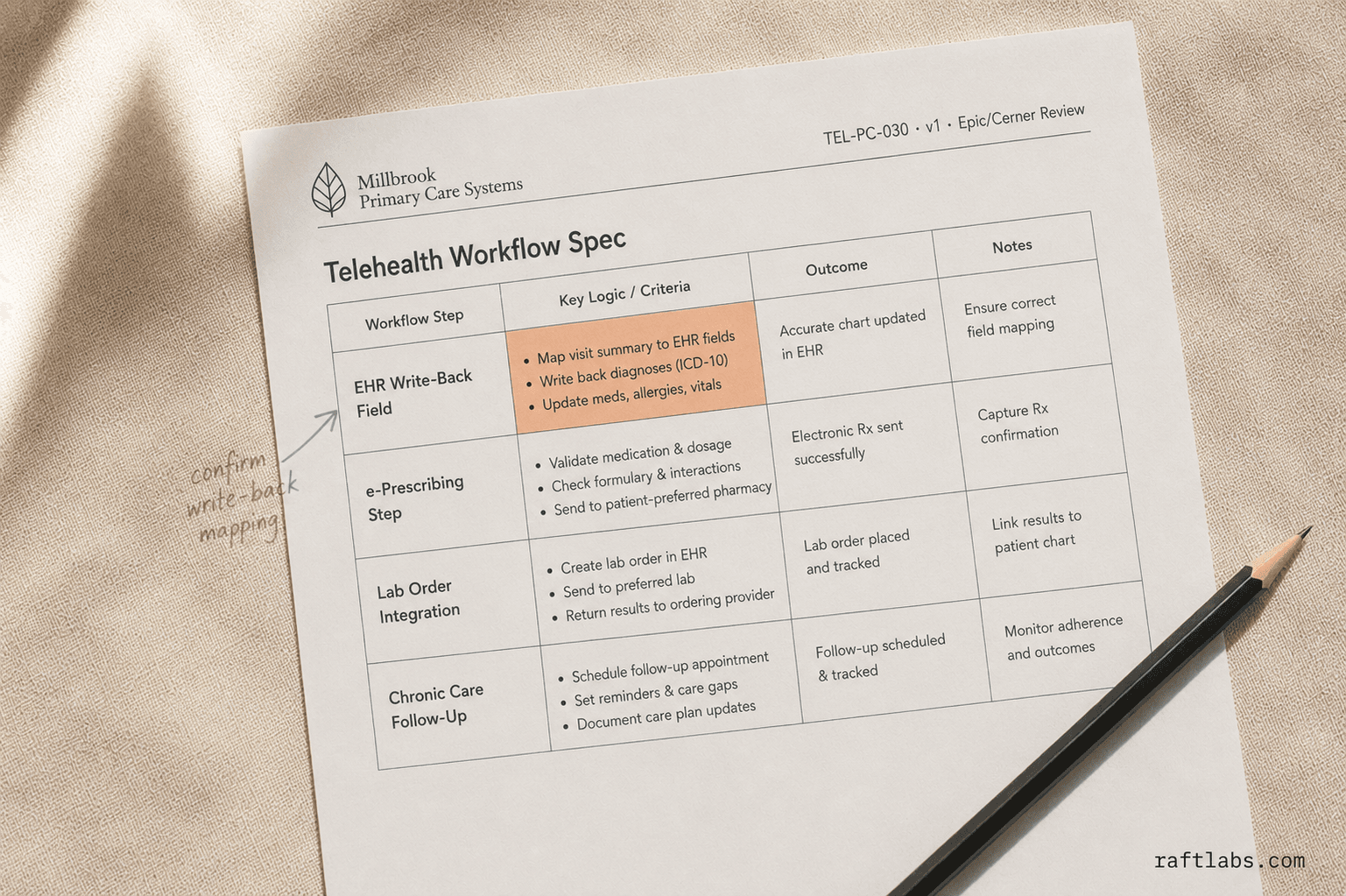
Service
Primary Care Telehealth Platform Development
See the service
Service
Women's Health App Development
See the serviceProof
Bella Skin Institute runs a fully automated loyalty program with no vendor lock-in
Read the case studyTry it yourself
HIPAA Compliance Checklist
30+ requirements, plain-English, filtered to what you're building.
Open the free tool