Pharmaceutical Software Development: Types, Cost, and Regulatory Requirements in 2026

Pharmaceutical software development is the design, build, validation, and maintenance of software used in drug manufacturing, clinical trials, quality management, and regulatory submissions. Systems range from LIMS ($80,000-$300,000) and QMS ($60,000-$200,000) to clinical trial management systems ($150,000-$500,000). All pharma software that touches product quality or patient safety must comply with FDA 21 CFR Part 11, GxP guidelines, and pass Computer System Validation (CSV). RaftLabs builds validated pharma software for US, UK, and Australian clients across clinical, manufacturing, and regulatory use cases.
Key Takeaways
- FDA 21 CFR Part 11 compliance is not optional for electronic records and signatures in pharma -- non-compliance has resulted in warning letters, import alerts, and facility shutdowns affecting hundreds of companies since 2020.
- Custom pharmaceutical software costs $50,000 to $500,000+ depending on type. A LIMS runs $80,000-$300,000. A clinical trial management system runs $150,000-$500,000.
- Computer System Validation (CSV) adds 20-40% to development cost but is mandatory for any software that affects product quality, patient safety, or regulatory submissions.
- Off-the-shelf LIMS and QMS products solve 70% of standard workflows. Custom development wins when your processes are non-standard, your integration footprint is large, or your regulatory exposure is high.
- Development timelines run 6-18 months depending on system complexity and validation scope. Budget the validation phase as a first-class project deliverable, not an afterthought.
An FDA warning letter arrived at a mid-size contract manufacturer in 2023. The reason: data integrity failures. Specifically, electronic records modified without audit trails, batch records completed on paper then re-entered into a system with no change history, and no way to prove that what was submitted to the FDA matched what actually happened on the production floor.
The company had software. It just wasn't pharma software. And nobody had validated it.
This guide is for the operations VP, clinical IT director, or biotech founder who knows something needs to change. It covers what pharmaceutical software actually is, what types of systems the industry runs on, what FDA and GxP compliance actually requires of your software, and what it costs to build it right.
TL;DR
The bottom line: Pharmaceutical software must be validated, auditable, and compliant with FDA 21 CFR Part 11 and GxP guidelines before it touches product quality or patient safety data.
| System Type | Custom Build Cost | Timeline |
|---|---|---|
| Laboratory Information Management System (LIMS) | $80,000–$300,000 | 6–12 months |
| Clinical Trial Management System (CTMS) | $150,000–$500,000 | 9–18 months |
| Quality Management System (QMS) | $60,000–$200,000 | 5–10 months |
| Regulatory Submission Platform | $100,000–$400,000 | 8–15 months |
| Pharmacovigilance System | $80,000–$250,000 | 6–12 months |
Validation (CSV/IQ/OQ/PQ) adds 20–40% to every figure above. Build that in from day one.
What is pharmaceutical software development?

Pharmaceutical software development is the design, build, validation, and ongoing maintenance of software systems used across the drug development and manufacturing lifecycle. It covers everything from the laboratory bench (LIMS, ELN) to the factory floor (MES, batch records) to the regulatory dossier (eCTD, eDMS).
What separates it from standard software development is the regulatory layer. Every system that creates, modifies, maintains, archives, retrieves, or transmits electronic records used in FDA submissions must comply with 21 CFR Part 11. Every system that affects product quality or patient safety must be validated. That validation is documented, auditable, and subject to inspection.
Standard software engineers know how to build features. Pharma software engineers know how to build features that survive a regulatory audit. The gap between those two things is where most pharma software projects fail.
Types of pharmaceutical software (and what each one does)
Most pharma and biotech operations depend on several interconnected systems. Here is what each one does and why it matters.
| System | Primary Function | Key Regulatory Concern |
|---|---|---|
| LIMS (Laboratory Information Management System) | Sample tracking, test results, QC workflows | Data integrity, 21 CFR Part 11, audit trails |
| CTMS (Clinical Trial Management System) | Trial planning, site management, patient enrollment | ICH E6 GCP, FDA 21 CFR Part 11 |
| QMS (Quality Management System) | SOPs, deviations, CAPAs, change control | GMP, ISO 9001, 21 CFR Part 820 |
| eDMS / eCTD Platform | Regulatory submissions and document management | FDA, EMA, ICH M4 |
| MES (Manufacturing Execution System) | Electronic batch records, production tracking | GMP, 21 CFR Part 211 |
| Pharmacovigilance System | Adverse event reporting, safety signal detection | ICH E2E, FDA 21 CFR Part 314 |
| Drug Track and Trace | Serialization, supply chain verification | DSCSA (US), FMD (EU) |
LIMS
A laboratory information management system tracks samples from receipt to result. It records who tested what, when, with which instrument, using which method. In a manual lab, that information lives in paper binders and shared spreadsheets. In a regulated lab, it must live in a validated system with a complete audit trail.
Without a validated LIMS, you cannot prove that a test result is original, unmodified, and was generated by the person who signed it. That is a 21 CFR Part 11 violation waiting to happen.
CTMS
A clinical trial management system manages the operational complexity of a clinical study: site activation, regulatory approvals, patient enrollment, protocol deviations, data queries, and sponsor-site communication. Modern CTMS platforms also connect to electronic data capture (EDC) systems that feed clinical data directly into the trial database.
The regulatory exposure here is significant. ICH E6(R3), the Good Clinical Practice guideline, requires that sponsors maintain adequate oversight of their trials. A CTMS that cannot produce a complete audit trail of site activity, protocol deviations, or adverse events is a compliance liability during an FDA inspection.
QMS
A quality management system is the backbone of GMP operations. It manages the documents that define how work is done (SOPs), the records that prove it was done correctly (batch records, test logs), and the processes that respond when something goes wrong (deviations, CAPAs, change controls).
For medical device companies, 21 CFR Part 820 (Quality System Regulation) mandates a design control process that a QMS must support. For drug manufacturers, 21 CFR Part 211 sets the GMP baseline.
Pharmacovigilance
Pharmacovigilance software collects, evaluates, and reports adverse events from marketed drugs. In the US, sponsors must submit Individual Case Safety Reports (ICSRs) to FDA MedWatch within 15 days for serious unexpected adverse events. In the UK, the MHRA requires SUSAR reporting within 7 days for fatal or life-threatening events. A pharmacovigilance system that cannot meet those timelines creates legal and regulatory exposure.
Regulatory requirements for pharma software
FDA 21 CFR Part 11
21 CFR Part 11 is the core US regulation for electronic records and electronic signatures. It applies to any system where electronic records replace paper records submitted to the FDA. The requirements include:
Audit trails that capture all record creation, modification, and deletion with user identity and timestamp
User authentication controls (unique user IDs, passwords, biometrics)
System validation proving the software does what it is designed to do
Access controls that limit what each user can view and modify
Secure, time-stamped electronic signatures with meaning (the equivalent of wet ink)
Non-compliance is not theoretical. The FDA has issued warning letters citing 21 CFR Part 11 violations for audit trail failures, shared user credentials, and lack of system validation. Those letters are public and have led to import alerts, consent decrees, and facility shutdowns.
GxP (Good Practice) guidelines
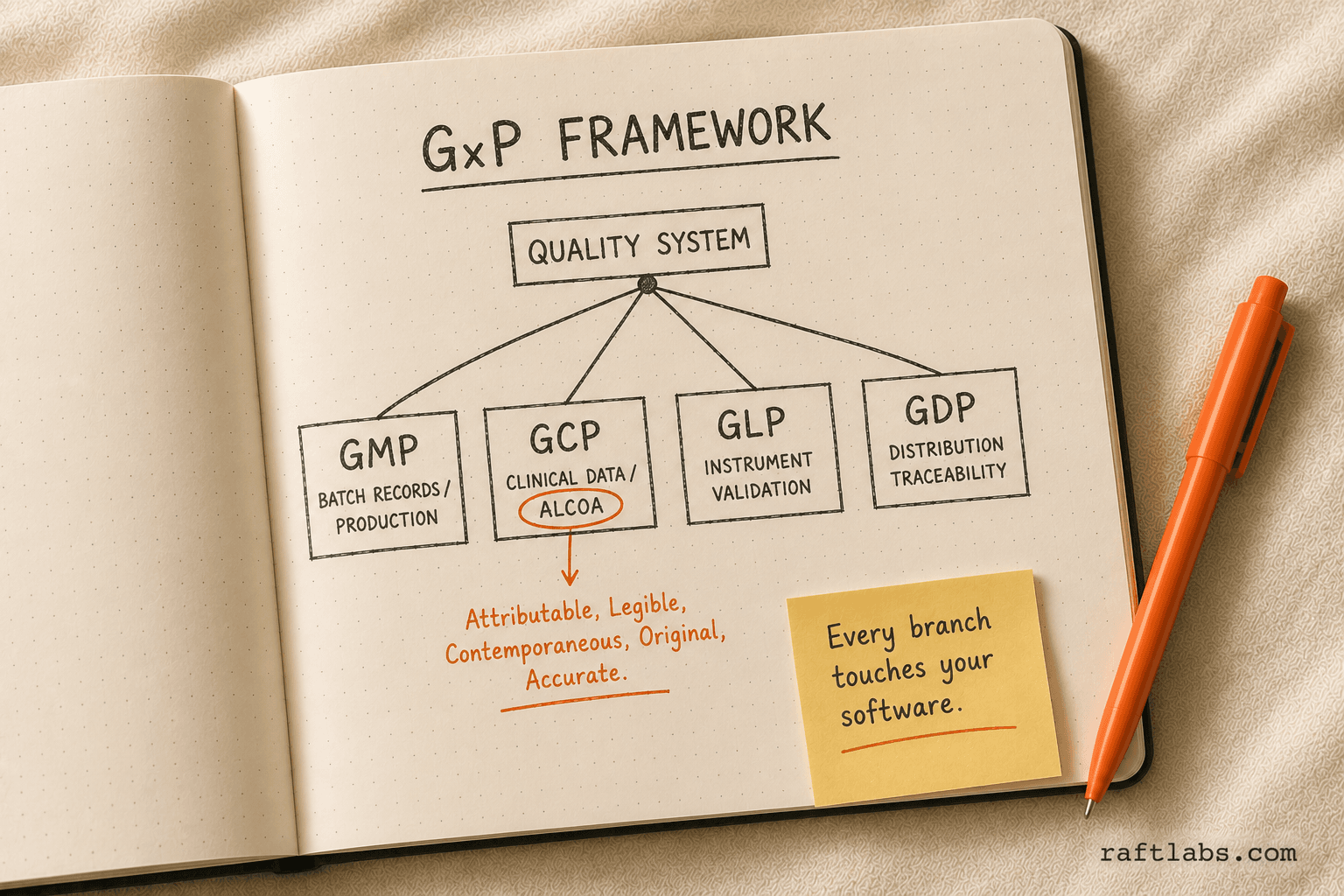
GxP is a shorthand for a family of quality guidelines: GMP (Good Manufacturing Practice), GCP (Good Clinical Practice), GLP (Good Laboratory Practice), and GDP (Good Distribution Practice). Each one applies to a different part of the pharma operation, and each one has software implications.
GMP requires electronic batch records to be complete, accurate, and accessible. Paper or legacy systems that allow backdating, overwriting, or gaps in data are GMP failures.
GCP requires that clinical data be attributable, legible, contemporaneous, original, and accurate. This is the ALCOA framework. Any software used in clinical data collection must meet these standards.
GLP applies to non-clinical safety studies. Instruments and the software that captures their data must be validated and calibrated.
EMA and international requirements
For companies operating in the EU, the EMA's GMP Annex 11 (Computerised Systems) is the equivalent of 21 CFR Part 11. It requires validation, change control, data integrity, and business continuity planning for all computerized systems used in GMP operations.
For Australia, the TGA follows ICH guidelines and the EU GMP framework. For Canada, Health Canada aligns with PIC/S guidance, which mirrors the EU annex.
Build vs buy in pharma: when custom wins
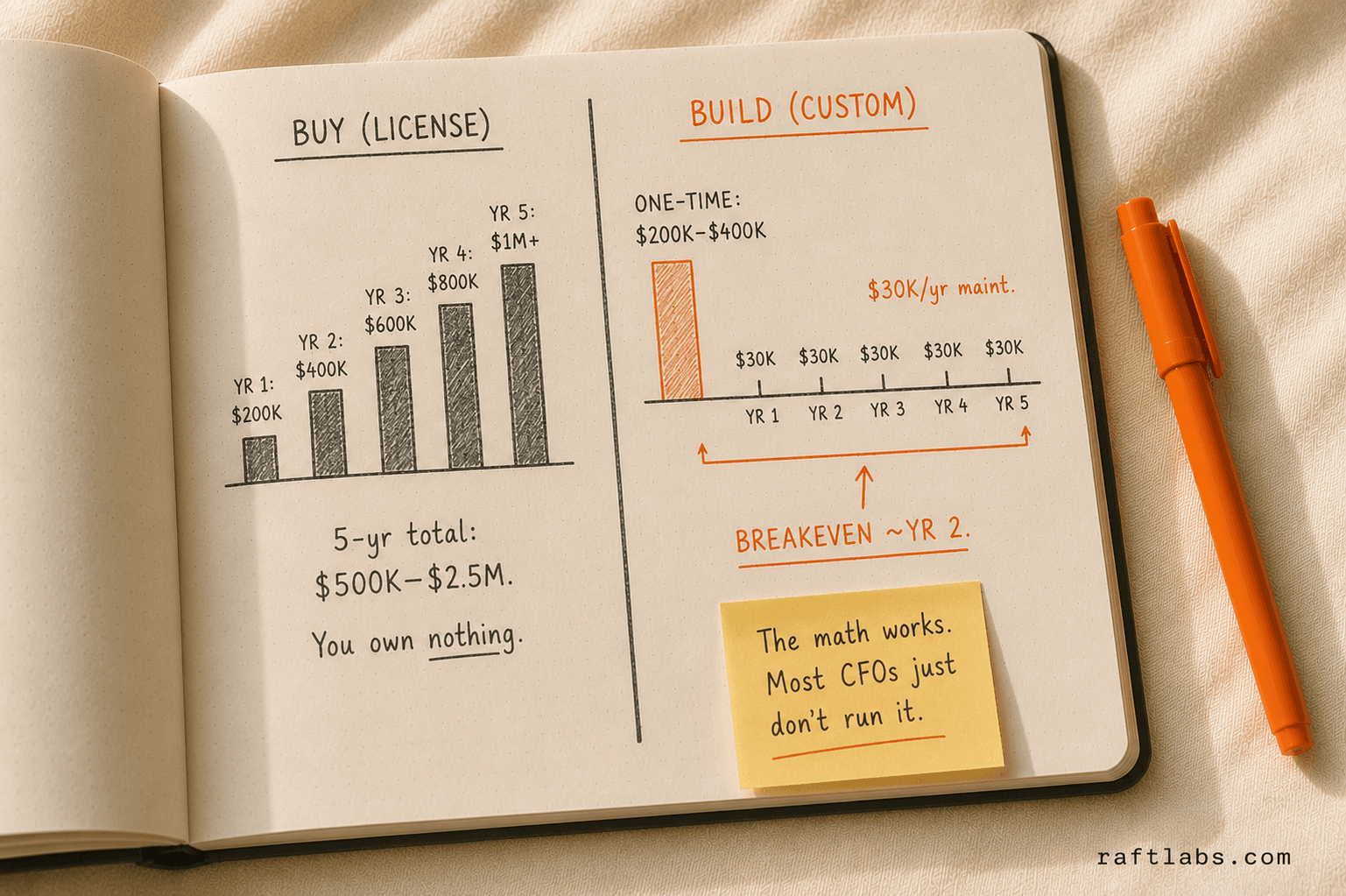
The pharma market has mature commercial products in every category. Veeva Vault dominates quality and regulatory. LabVantage and STARLIMS are leading LIMS platforms. Medidata Rave is widely used for EDC in clinical trials.
Off-the-shelf wins when your processes are standard. If you run clinical trials the same way most sponsors do, Medidata Rave solves your problem. If your QMS needs match what Veeva's template library covers, you do not need a custom build.
Custom development wins in these situations:
Non-standard processes. Some pharma operations have proprietary manufacturing processes, unusual sample workflows, or specialized analytical methods that commercial products cannot configure to match. Forcing your process into a commercial product's data model creates manual workarounds. Those workarounds become compliance liabilities.
Deep integration requirements. A LIMS that must talk to a 20-year-old ERP, a proprietary instrument management system, and a bespoke MES requires integration work that commercial vendors often price separately at enterprise rates. Custom-built integration is often cheaper and more maintainable.
Audit trail and data integrity controls. Commercial products have audit trails. Custom builds let you define exactly what gets logged, at what granularity, in what format. For organizations under heightened FDA scrutiny, that control matters.
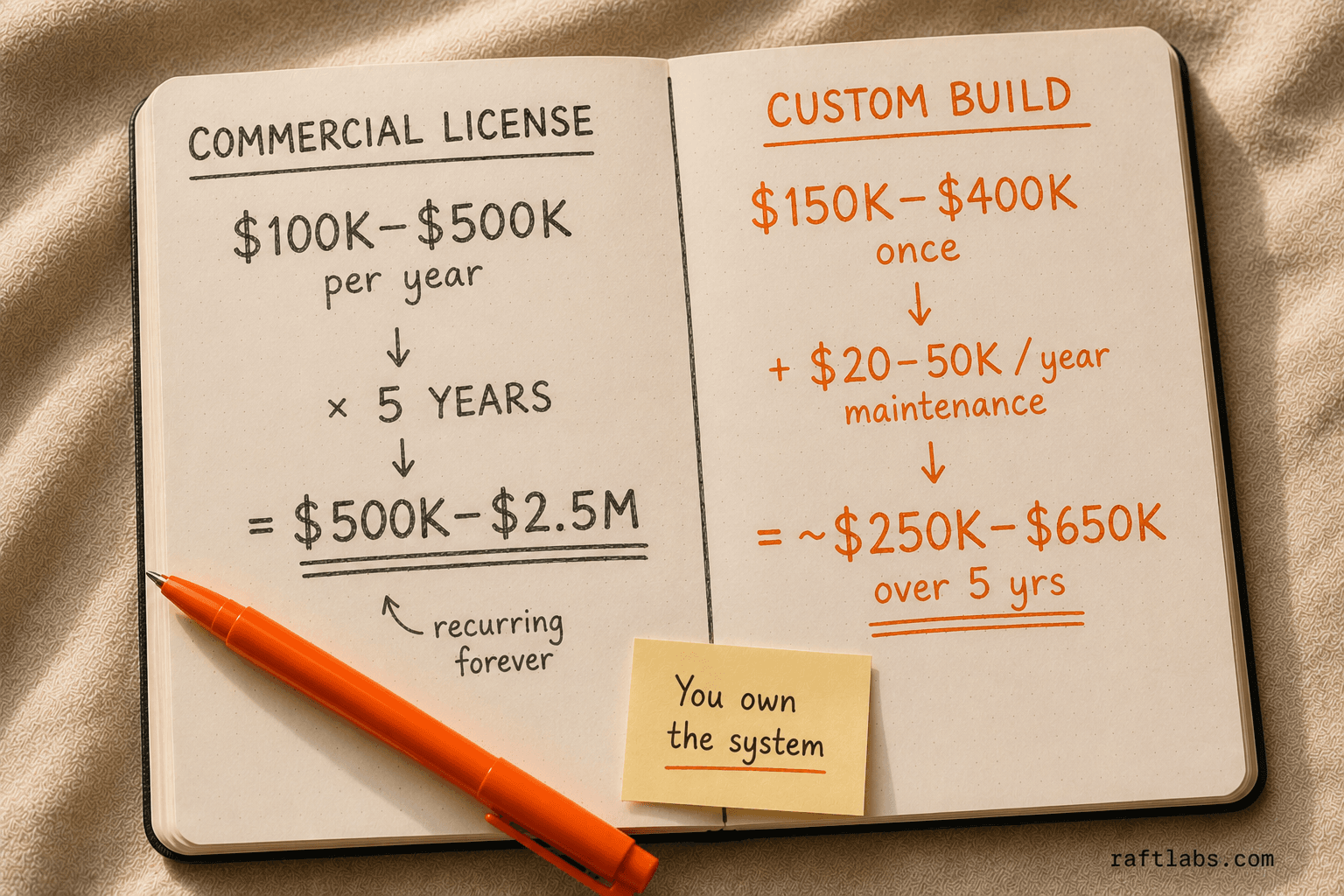
Total cost of ownership. Enterprise pharma software licenses run $100,000-$500,000+ per year. At 5 years, you have spent $500,000-$2.5M in licensing alone. A custom build at $150,000-$400,000 with $20,000-$50,000/year in maintenance often has a lower 5-year TCO -- and you own the system.
Pharmaceutical software development cost
Cost ranges by system type, based on custom development for US/UK/AU-based compliance requirements:
| System | Base Build | Validation (CSV) | Total Range |
|---|---|---|---|
| LIMS | $80,000–$200,000 | $20,000–$80,000 | $100,000–$280,000 |
| CTMS | $150,000–$350,000 | $40,000–$120,000 | $190,000–$470,000 |
| QMS | $60,000–$150,000 | $15,000–$60,000 | $75,000–$210,000 |
| eDMS / Regulatory Submission | $100,000–$300,000 | $25,000–$90,000 | $125,000–$390,000 |
| Pharmacovigilance | $80,000–$200,000 | $20,000–$70,000 | $100,000–$270,000 |
| MES / Electronic Batch Records | $100,000–$300,000 | $30,000–$100,000 | $130,000–$400,000 |
What drives cost up:
Complex integration with legacy ERP or instrument systems ($20,000-$80,000 per integration)
Multi-site or multi-tenant requirements (add 30-50%)
21 CFR Part 11 electronic signature workflows with biometric components
Compliance with multiple regulatory frameworks simultaneously (FDA + EMA + TGA)
What keeps cost down:
Well-documented, stable processes before development starts
Phased scope: core system first, integrations in later releases
Development team with prior pharma validation experience (validation rework is expensive)
Validation requirements: IQ, OQ, PQ, and CSV
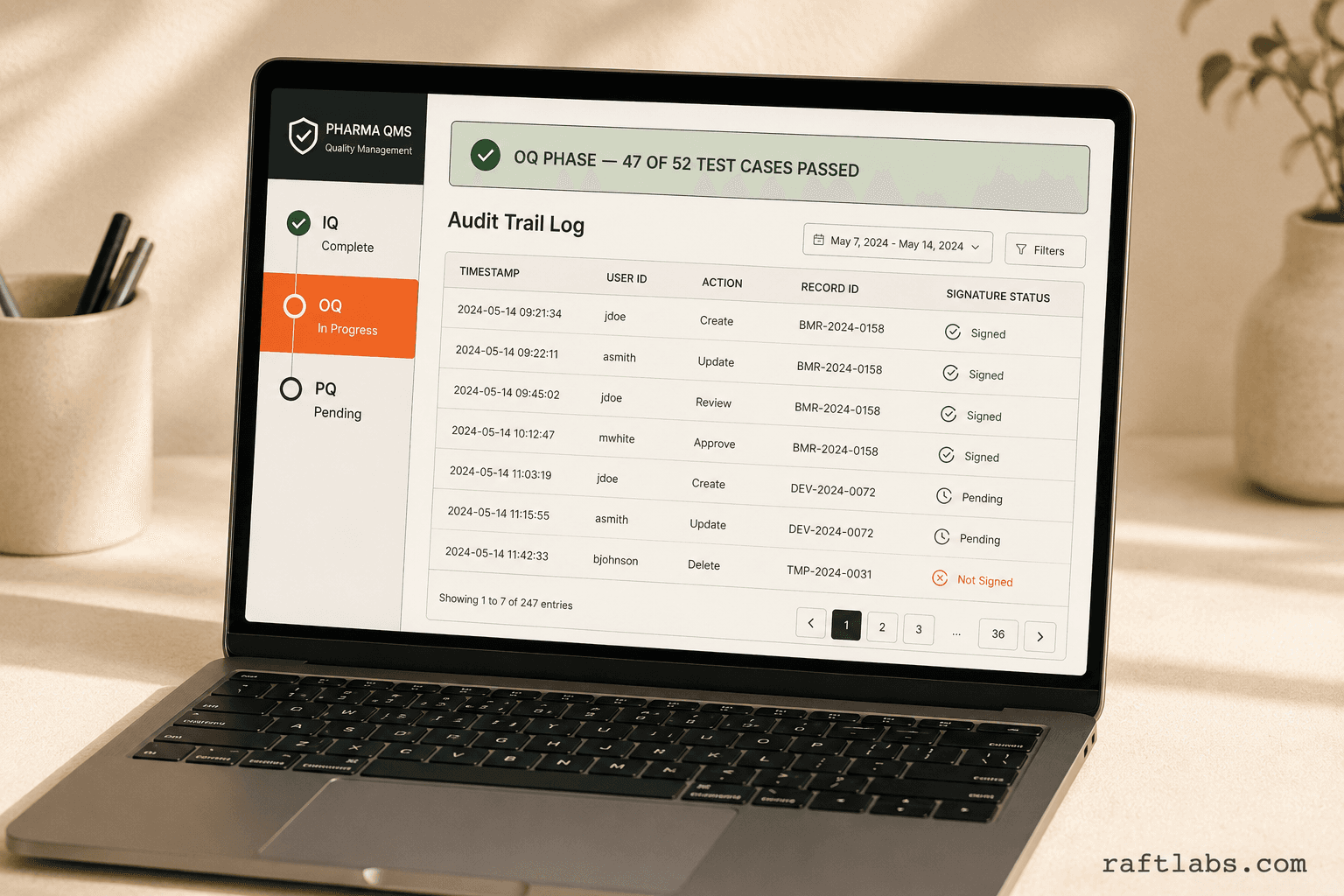
Computer System Validation (CSV) is not an optional add-on. It is mandatory for any software system that affects product quality, patient safety, or regulated data.

The FDA's 21 CFR Part 11 guidance and the ISPE GAMP 5 framework define the validation approach:
Installation Qualification (IQ). Documents that the system is installed correctly in the intended environment. Server specs, software versions, network configuration -- all documented and signed off. This phase typically runs 2-4 weeks.
Operational Qualification (OQ). Tests that the system functions as specified. Each requirement in the design specification gets a test case. Pass/fail recorded. Deviations documented and resolved. This phase takes 4-8 weeks for a mid-complexity system.
Performance Qualification (PQ). Tests that the system performs correctly in the actual operational environment with real users and representative data. This is the closest thing to a dress rehearsal before go-live. Typically 4-8 weeks.
Ongoing validation. Every significant change to the system after go-live triggers a revalidation cycle. Change control is the process that governs this. It is not a project management overhead -- it is a regulatory requirement.
A pharma software project that skips validation is not complete. It is undeployable in a regulated environment. Budget it from day one.
Development timeline
Realistic timelines for custom pharmaceutical software, including validation:
| Phase | Duration |
|---|---|
| Discovery and requirements (URS, FRS) | 4–8 weeks |
| Architecture and design | 3–6 weeks |
| Development (iterative sprints) | 12–24 weeks |
| System integration testing | 4–8 weeks |
| IQ/OQ (Installation + Operational Qualification) | 6–12 weeks |
| PQ (Performance Qualification) | 4–8 weeks |
| User acceptance testing | 2–4 weeks |
| Go-live and hypercare | 2–4 weeks |
Total: 37–74 weeks (approximately 9–18 months) for a full-scope system.
The timeline compression that works in commercial software does not apply here. Skipping the user requirements specification (URS) creates ambiguity that shows up during OQ as failed test cases, which require deviation reports, root cause analysis, and retest cycles. Each retest cycle adds weeks.
The fastest way through pharma software validation is to do it right the first time.
Tech stack and integration requirements
Architecture considerations
Pharma software runs on the same technology stacks as other enterprise software: Python, Java, .NET, Node.js, React, PostgreSQL, AWS, Azure. The technology is not special. What is special is how it is used.
Audit trails require database-level logging -- not application-level. A trigger-based audit table captures every insert, update, and delete with user identity and timestamp. Application-level logging can be bypassed; database-level logging cannot.
Access control requires role-based permissions that are granular enough to satisfy 21 CFR Part 11 requirements. Not just "admin vs read-only" -- the system must distinguish who can create a record, who can review it, who can approve it, and who can never see it at all.
Electronic signatures require the system to capture the meaning of the signature (the equivalent of "I approved this batch record") alongside the identity and timestamp of the signer.
Common integration requirements
Most pharma software projects involve at least one of these integrations:
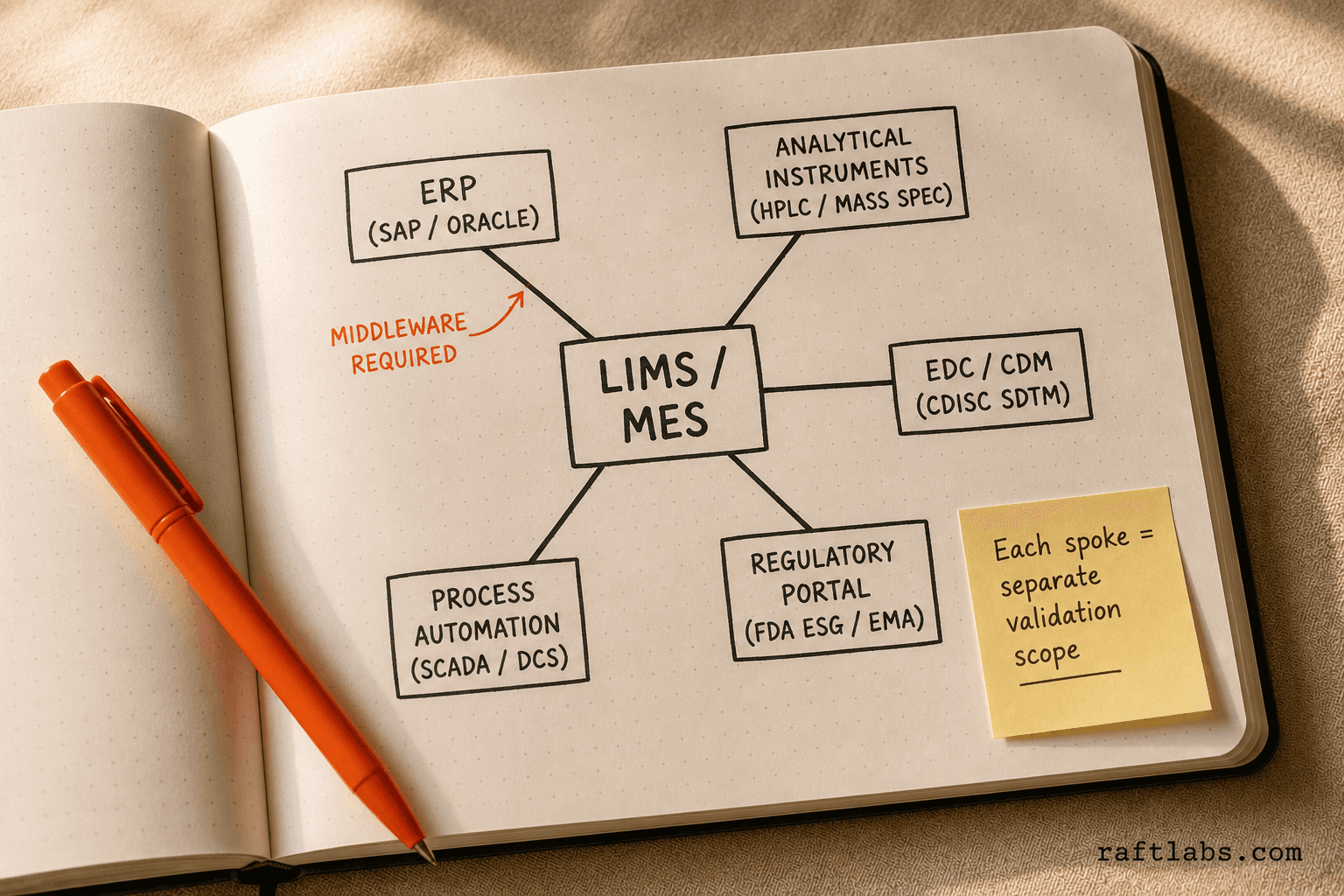
ERP systems: SAP, Oracle, or Microsoft Dynamics for production planning, inventory, and procurement. The LIMS or MES must pull sample data and push results back to the ERP.
Instrument management: LIMS must connect to analytical instruments (HPLC, mass spectrometers, dissolution testers) via direct interface or an instrument data management system (IDMS).
EDC/CDM: Clinical data management systems must export data in CDISC SDTM format for FDA submissions.
Regulatory portals: eCTD submissions require integration with FDA's Electronic Submissions Gateway and EMA's XEVMPD or EVWEB.
MES/EBR: Electronic batch record systems must connect to process automation (SCADA, DCS) to capture in-process data automatically rather than via manual entry.
Each integration is a separate scope item. Budget it, validate it, and test the failure modes -- what happens when the ERP is unavailable, what happens when the instrument connection drops.
What to look for in a pharmaceutical software development partner
Most software development firms can build a CRUD application. Fewer can build one that passes an FDA audit. The difference is not about technology -- it is about experience with validated systems.
Ask any prospective partner these questions:
Have you built software that has been inspected by FDA or EMA? Can you share the outcome?
What is your standard approach to user requirements specifications and design specifications?
Who owns the validation documentation: your team or the client?
Have you worked with GAMP 5? What category does the software you build typically fall under?
What change control process do you follow for post-launch modifications?
A development firm that cannot answer those questions specifically is not a pharma software firm. They are a general software firm quoting a pharma project. That is a risk you do not want to take when the audit trails you build are the evidence the FDA inspects.
RaftLabs builds validated pharmaceutical software for US, UK, and Australian clients. Every engagement starts with a scoping session that covers regulatory requirements before a line of code is written. If you are scoping a LIMS, QMS, CTMS, or custom regulatory platform, start the conversation here.
Frequently asked questions
What is pharmaceutical software development?
Pharmaceutical software development is the process of designing, building, validating, and maintaining software systems used in drug discovery, clinical trials, manufacturing, quality management, and regulatory submissions. It differs from standard software development because every system that touches product quality or patient safety must comply with FDA 21 CFR Part 11, GxP guidelines, and pass a formal Computer System Validation (CSV) process before deployment.
How much does pharmaceutical software cost to develop?
A LIMS costs $100,000-$280,000 including validation. A CTMS costs $190,000-$470,000. A QMS costs $75,000-$210,000. A regulatory submission platform costs $125,000-$390,000. Pharmacovigilance systems cost $100,000-$270,000. Validation (CSV/IQ/OQ/PQ) adds 20-40% to every category and is mandatory for systems that affect product quality or patient safety.
What is FDA 21 CFR Part 11 and why does it matter for software?
FDA 21 CFR Part 11 is the federal regulation governing electronic records and electronic signatures in pharmaceutical and biotech companies. Any software system that creates, modifies, maintains, archives, retrieves, or transmits electronic records used in FDA submissions must comply. Non-compliance has led to FDA warning letters, import alerts, and consent decrees. The regulation covers audit trails, access controls, data integrity, and e-signature authentication.
What is Computer System Validation (CSV) in pharma software?
Computer System Validation (CSV) is a documented process proving a software system consistently does what it is designed to do. It includes Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ). CSV is required by FDA 21 CFR Part 11 and GxP guidelines for any system affecting product quality or patient safety. Skipping it makes the system non-deployable in a regulated environment.
When should a pharma company build custom software instead of buying off-the-shelf?
Build custom when your processes deviate significantly from commercial LIMS or QMS products, when you need deep integration with legacy ERP or MES systems that commercial vendors don't support natively, when your regulatory exposure requires audit trail and data integrity controls the commercial product cannot provide, or when per-seat licensing at scale exceeds the amortized cost of a custom build over 3-5 years.
Frequently asked questions
- Pharmaceutical software development is the process of designing, building, validating, and maintaining software systems used in drug discovery, clinical trials, manufacturing, quality management, and regulatory submissions. It differs from standard software development because every system that touches product quality or patient safety must comply with FDA 21 CFR Part 11, GxP guidelines, and pass a formal Computer System Validation (CSV) process before deployment.
- A laboratory information management system (LIMS) costs $80,000-$300,000 to develop custom. A clinical trial management system (CTMS) runs $150,000-$500,000. A quality management system (QMS) runs $60,000-$200,000. A regulatory submission platform runs $100,000-$400,000. Pharmacovigilance systems run $80,000-$250,000. Validation (CSV/IQ/OQ/PQ) adds 20-40% to every category.
- FDA 21 CFR Part 11 is the federal regulation governing electronic records and electronic signatures in pharmaceutical and biotech companies. Any software system that creates, modifies, maintains, archives, retrieves, or transmits electronic records used in FDA submissions must comply. Non-compliance has led to FDA warning letters, import alerts, and consent decrees. The regulation covers audit trails, access controls, data integrity, and e-signature authentication.
- Computer System Validation (CSV) is a documented process that proves a software system consistently does what it is designed to do. It includes Installation Qualification (IQ), which confirms the system is installed correctly; Operational Qualification (OQ), which confirms the system functions as specified; and Performance Qualification (PQ), which confirms the system performs correctly in the actual operational environment. CSV is required by FDA 21 CFR Part 11 and GxP guidelines for any system affecting product quality or patient safety.
- Build custom when your processes deviate significantly from what commercial LIMS or QMS products support, when you need deep integration with legacy ERP or MES systems that commercial vendors don't support natively, when your regulatory exposure (FDA, EMA, TGA) requires audit trail and data integrity controls the commercial product cannot provide, or when your per-seat licensing cost at scale exceeds the amortized cost of a custom build over 3-5 years.
Ask an AI
Get an instant summary of this post from your preferred AI assistant.
Related articles

How to Build an App Like Blinkit: Quick Commerce Architecture, Dark Store Ops, and Real Build Costs
Learn what it takes to build a hyperlocal quick commerce platform like Blinkit, features, costs, and how RaftLabs helps you launch faster.

How to Build an App Like Grubhub: A Founder's Guide to Restaurant Discovery and Delivery Platforms
For campus dining operators, restaurant co-ops, and city-specific delivery platforms, this guide breaks down the real cost, timeline, and operational complexity of building a Grubhub alternative — and when it pays to stop paying commissions.
How to Build a Community Platform Like Circle.so: Architecture for Membership Businesses
Circle.so charges communities up to $399 per month plus a 4% transaction fee. For media companies, professional associations, and B2B SaaS teams with 1,000+ members, that math inverts fast. Here is how to build a branded community platform, what it costs, and when custom wins.